









Përmbajtje
Sëmundja Recklinghausen
Cfare eshte
Sëmundja e Recklinghausen quhet edhe neurofibromatoza e tipit I.
Termi "neurofibromatozë" përfshin një sërë sëmundjesh gjenetike që ndikojnë në zhvillimin qelizor të indeve neuronale. Ekzistojnë dy lloje të neurofibromatozës: tipi I dhe tipi II. Këto dy forma, megjithatë, kanë karakteristika të ngjashme dhe shkaktohen nga mutacione në gjene të ndryshme.
Neurofibromatoza e tipit I është displazi neurodermale, një anomali në zhvillimin e indit neuronal. Kjo patologji u përshkrua për herë të parë në 1882 nga Friederich Daniel Von Recklinghausen, prej nga vjen emri aktual i kësaj patologjie.
Ndryshimet në indin neuronal shfaqen nga zhvillimi embrional.
Neurofibromatoza e tipit I është forma më e zakonshme e neurofibromatozës me 90% të rasteve të tipit I. Është gjithashtu një nga sëmundjet gjenetike më të zakonshme të njeriut me prevalencë (numri i rasteve në një popullatë të caktuar, në një kohë të caktuar) që arrin në 1/ 3 lindje. Për më tepër, nuk u vu re asnjë mbizotërim midis burrave dhe grave. (000)
Sëmundja Recklinghausen është një sëmundje gjenetike e trashëguar në të cilën mënyra e transmetimit është autosomike dominuese. Ose, që prek një kromozom jo seksual dhe për të cilin prania e vetëm njërës nga dy kopjet e gjenit të mutuar është e mjaftueshme që subjekti të zhvillojë sëmundjen. Kjo sëmundje është pasojë e ndryshimeve në gjenin NF1 që ndodhet në kromozomin 17q11.2.
Karakteristikat e sëmundjes përcaktohen përmes: (2)
- butona me ngjyra “café-au-lait”;
– glioma optike (tumore në nivelin e rrënjëve të nervit okular);
– Nyjet lish (hematoma që pigmentojnë irisin e syve);
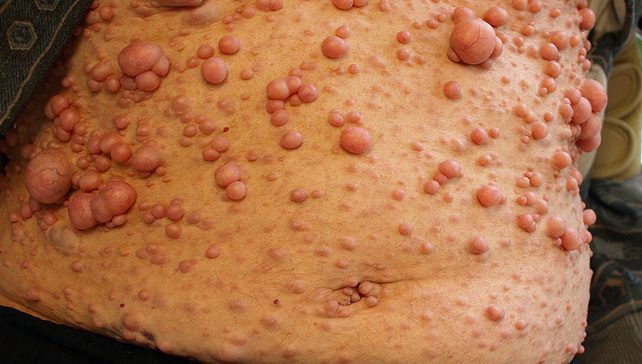
– neurofibroma të nervave kurrizore dhe periferike;
– dëmtim neurologjik dhe/ose njohës;
– skolioza;
– anomalitë e fytyrës;
– tumoret malinje të mbështjellësit nervor;
– feokromocitoma (tumor malinj i lokalizuar në veshka);
– lezione kockore.
Simptomat
Sëmundja Recklinghausen prek lëkurën dhe sistemin nervor qendror dhe periferik. Simptomat primare të shoqëruara zakonisht shfaqen në fëmijëri dhe mund të prekin lëkurën si më poshtë: (4)
– Njollat me ngjyrë të lëkurës “café au lait”, të madhësive të ndryshme, të formave të ndryshme dhe që mund të gjenden në çdo nivel të trupit;
– njollat që zhvillohen nën krahë dhe në sqetull;
– zhvillimi i tumoreve në nervat periferikë;
– zhvillimi i tumoreve në rrjetin nervor.
Shenjat dhe simptomat e tjera mund të jenë gjithashtu domethënëse të sëmundjes, këto përfshijnë:
– Nyjet lish: rritja që prek sytë;
– një feokromocitoma: tumor i gjëndrës mbiveshkore, nga të cilat dhjetë për qind e këtyre tumoreve janë kancerogjene;
– zmadhimi i mëlçisë;
– një glioma: tumor i nervit optik.
Ndikimi i sëmundjes në zhvillimin e kockave përfshin ndërtimin e shkurtër, deformimet e kockave dhe skoliozën. (4)
Origjina e sëmundjes
Sëmundja Recklinghausen është një sëmundje gjenetike e trashëguar e formës autosomale dominante. Qoftë që ka të bëjë me një kromozom jo seksual dhe për të cilin prania e vetëm njërës nga dy kopjet e gjenit të mutuar është e mjaftueshme për zhvillimin e sëmundjes.
Sëmundja shkaktohet nga një sërë mutacionesh në gjenin NF1, i vendosur në kromozomin 17q11.2. Është një nga mutacionet spontane më të zakonshme në mesin e të gjitha sëmundjeve gjenetike njerëzore.
Vetëm 50% e pacientëve me gjenin NF1 të mutuar kanë një histori familjare të transmetimit të sëmundjes. Pjesa tjetër e pacientëve në fjalë kanë mutacione spontane në këtë gjen.
Shprehja e sëmundjes është shumë e ndryshueshme nga një individ në tjetrin me një panel manifestimesh klinike që mund të variojnë nga komplikime të lehta deri në shumë më të rënda. (2)
Faktorët e rrezikut
Faktorët e rrezikut për zhvillimin e sëmundjes janë gjenetikë.
Në të vërtetë, sëmundja transmetohet nga një transferim i gjenit të mutuar NF1 sipas mënyrës autosomale dominuese.
Ose mutacioni në fjalë ka të bëjë me një gjen të vendosur në një kromozom jo seksual. Përveç kësaj, prania e vetëm njërës nga dy kopjet e gjenit të mutuar është e mjaftueshme që sëmundja të zhvillohet. Në këtë kuptim, një individ prej të cilit njëri prej prindërve ka fenotipin e sëmundjes ka një rrezik 50% që ta zhvillojë vetë patologjinë.
Parandalimi dhe trajtimi
Diagnoza e sëmundjes është para së gjithash diferenciale, veçanërisht në lidhje me praninë e disa simptomave karakteristike. Objektivi kryesor i mjekut është të përjashtojë të gjitha mundësitë e sëmundjeve të tjera të përfshira në këto manifestime klinike.
Këto sëmundje, simptomat e të cilave ngjajnë shumë me ato të sëmundjes së Recklinghausen, përfshijnë:
– Sindroma e leopardit: një sëmundje gjenetike, simptomat e së cilës mbulojnë edhe njollat kafe në lëkurë, zmadhimin e hapësirës midis syve, ngushtimin e arteries koronare, humbjen e dëgjimit, një ndërtim të vogël dhe anomalitë në sinjalet elektrike të zemrës;
– melanoma neurokutane: një sëmundje gjenetike që shkakton zhvillimin e qelizave tumorale në tru dhe palcën kurrizore;
– Schwannomatoza, një sëmundje e rrallë që shkakton zhvillimin e tumoreve në indin nervor;
– Sindroma Watson: një sëmundje gjenetike që çon gjithashtu në zhvillimin e nyjeve të Lish-it, një ndërtim të vogël, neurofibroma, një kokë anormalisht të madhe dhe ngushtim të arteries pulmonare.
Ekzaminimet shtesë më pas bëjnë të mundur konfirmimin ose jo të sëmundjes, në veçanti ky është rasti i MRI (Magnetic Rezonance Imaging) apo edhe i skanerit. (4)
Në kontekstin e një sëmundjeje komplekse, trajtimi i saj duhet të administrohet në pjesë të ndryshme të trupit në fjalë.
Trajtimet e përshkruara në fëmijëri përfshijnë:
– vlerësimi i aftësive të të nxënit;
– vlerësimi i hiperaktivitetit të mundshëm;
– trajtimi i skoliozës dhe deformimeve të tjera të jashtëzakonshme.
Tumoret mund të trajtohen nga: (4)
– heqja laparoskopike e tumoreve kancerogjene;
– kirurgji për heqjen e tumoreve që prekin nervat;
– radioterapi;
– kimioterapia.