









Përmbajtje
Skleroza tuberoze Bourneville
Cfare eshte
Skleroza tuberoze e Bourneville është një sëmundje komplekse gjenetike e karakterizuar nga zhvillimi i një tumori beninj (jo kanceroz) në pjesë të ndryshme të trupit. Këto tumore më pas mund të lokalizohen në lëkurë, tru, veshka dhe organe dhe inde të tjera. Kjo patologji mund të shkaktojë edhe probleme serioze në zhvillimin e individit. Megjithatë, manifestimet klinike dhe ashpërsia e sëmundjes ndryshojnë nga pacienti në pacient.
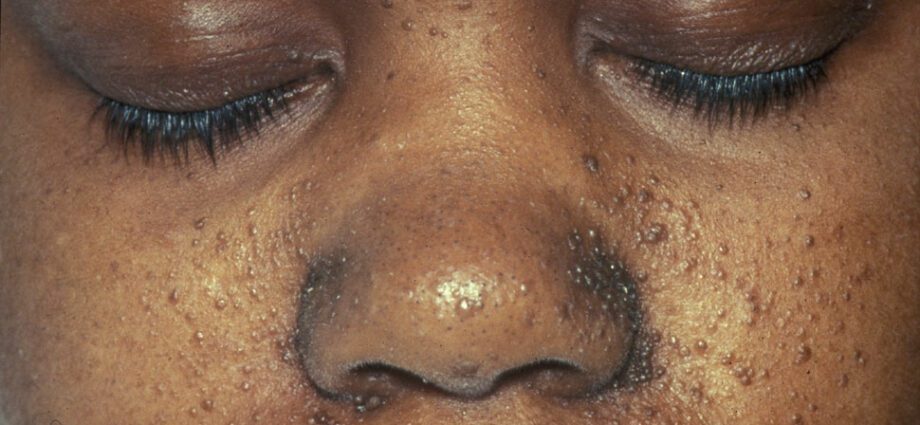
Anomalitë e lidhura të lëkurës janë përgjithësisht të ngjashme me njollat në lëkurë ose me zonat ku lëkura është më e lehtë se në pjesën tjetër të trupit. Zhvillimi i tumoreve në fytyrë quhet angiofibroma.
Në kuadrin e dëmtimit të trurit, shenjat klinike janë kriza epileptike, probleme të sjelljes (hiperaktivitet, agresivitet, paaftësi intelektuale, probleme në të mësuar etj.). Disa fëmijë me këtë sëmundje madje kanë një formë të autizmit, çrregullime zhvillimore, që ndikojnë në ndërveprimet sociale dhe komunikimin. Tumoret beninje të trurit mund të shkaktojnë gjithashtu komplikime që mund të jenë fatale për subjektin.
Zhvillimi i tumoreve në veshka është i zakonshëm tek njerëzit me sklerozë tuberoze. Kjo mund të shkaktojë komplikime të rënda në funksionin e veshkave. Përveç kësaj, tumoret mund të zhvillohen në zemër, mushkëri dhe retinë. (2)
Është një sëmundje e rrallë, prevalenca e së cilës (numri i rasteve në një popullatë të caktuar në një kohë të caktuar) shkon në 1/8 deri në 000/1 persona. (15)
Simptomat
Manifestimet klinike të lidhura me sklerozën tuberoze të Bourneville ndryshojnë sipas organeve të prekura. Përveç kësaj, simptomat e lidhura me sëmundjen ndryshojnë shumë nga një individ në tjetrin. Me simptoma që variojnë nga të lehta në të rënda.
Simptomat më të identifikuara të kësaj sëmundje përfshijnë krizat epileptike, çrregullimet njohëse dhe të sjelljes, anomalitë e lëkurës, etj. Organet që preken më shpesh janë: truri, zemra, veshkat, mushkëritë dhe lëkura.
Zhvillimi i tumoreve malinje (kancerogjene) është i mundshëm në këtë sëmundje, por janë të rrallë dhe prekin kryesisht veshkat.
Shenjat klinike të sëmundjes në tru vijnë nga sulmet në nivele të ndryshme:
– dëmtimi i tuberkulozit kortikal;
– nyjet ependimale (SEN);
– astrocitoma gjigante ependimale.
Ato rezultojnë me: zhvillim të prapambetjes mendore, vështirësi në të mësuar, çrregullime të sjelljes, agresivitet, çrregullime të vëmendjes, hiperaktivitet, çrregullime obsesive-kompulsive etj.
Dëmtimi i veshkave karakterizohet nga zhvillimi i cisteve ose angiomiolipomave. Këto mund të çojnë në dhimbje të veshkave dhe madje edhe në dështim të veshkave. Nëse vërehet gjakderdhje e rëndë, mund të jetë nga anemia e rëndë ose presioni i lartë i gjakut. Mund të jenë të dukshme edhe pasoja të tjera më të rënda, por të rralla, veçanërisht zhvillimi i karcinomave (tumori i qelizave përbërëse të epitelit).
Dëmtimi i syrit mund të jetë i ngjashëm me njollat e dukshme në retinë, duke shkaktuar shqetësime vizuale apo edhe verbëri.
Anomalitë e lëkurës janë të shumta:
– makulat hipomelanike: të cilat rezultojnë në shfaqjen e njollave të lehta në lëkurë, kudo në trup, si pasojë e mungesës së melaninës, një proteinë që i jep ngjyrë lëkurës;
– shfaqja e njollave të kuqe në fytyrë;
– njolla të zbardhura në ballë;
– anomali të tjera të lëkurës, të varura nga një individ në tjetrin.
Lezionet e mushkërive janë të pranishme në 1/3 e pacientëve me një mbizotërim të lehtë femëror. Simptomat shoqëruese janë më pas vështirësi pak a shumë të rënda në frymëmarrje.
Origjina e sëmundjes
Origjina e sëmundjes është gjenetike dhe e trashëgueshme.
Transmetimi përfshin mutacione në gjenet TSC1 dhe TSC2. Këto gjene me interes hyjnë në lojë në formimin e proteinave: hamartin dhe tuberin. Këto dy proteina bëjnë të mundur, nëpërmjet një loje ndërvepruese, rregullimin e proliferimit të qelizave.
Pacientët me këtë sëmundje lindin me të paktën një kopje të mutuar të këtyre gjeneve në secilën prej qelizave të tyre. Këto mutacione më pas kufizojnë formimin e hamartinës ose tubertinës.
Në kontekstin ku dy kopjet e gjenit janë të mutuara, ato parandalojnë plotësisht prodhimin e këtyre dy proteinave. Prandaj, kjo mungesë proteine nuk e lejon më trupin të rregullojë rritjen e qelizave të caktuara dhe, në këtë kuptim, çon në zhvillimin e qelizave tumorale në inde dhe/ose organe të ndryshme.
Faktorët e rrezikut
Faktorët e rrezikut për zhvillimin e një patologjie të tillë janë gjenetikë.
Në të vërtetë, transmetimi i sëmundjes është efektiv përmes një mënyre autosomale dominuese. Ose, gjeni i mutuar me interes ndodhet në një kromozom jo seksual. Përveç kësaj, prania e vetëm njërës nga dy kopjet e gjenit të mutuar është e mjaftueshme që sëmundja të zhvillohet.
Në këtë kuptim, një individ që ka njërin prej këtyre dy prindërve që vuan nga sëmundja ka një rrezik 50% për të zhvilluar vetë fenotipin e sëmurë.
Parandalimi dhe trajtimi
Diagnoza e sëmundjes është para së gjithash diferenciale. Ai bazohet në kritere fizike atipike. Në shumicën e rasteve, shenjat e para karakteristike të sëmundjes janë: prania e krizave të përsëritura epileptike dhe vonesa në zhvillimin e subjektit. Në raste të tjera, këto shenja të para rezultojnë me njolla të lëkurës ose identifikimin e një tumori në zemër.
Pas kësaj diagnoze të parë, ekzaminimet shtesë janë thelbësore për të vërtetuar diagnozën ose jo. Kjo perfshin:
– një skanim i trurit;
– një MRI (Rezonancë Magnetike) e trurit;
– një ekografi e zemrës, mëlçisë dhe veshkave.
Diagnoza mund të jetë efektive në lindjen e fëmijës. Përndryshe, është e rëndësishme që ajo të kryhet sa më shpejt që të jetë e mundur për të marrë në dorë pacientin sa më shpejt të jetë e mundur.
Aktualisht, nuk ka kurë për sëmundjen. Prandaj, trajtimet shoqëruese janë të pavarura nga simptomat e paraqitura nga secili individ.
Zakonisht, ilaçet anti-epileptike jepen për të kufizuar krizat. Përveç kësaj, janë të përshkruara edhe barna për trajtimin e qelizave tumorale të trurit dhe veshkave. Në kuadrin e problemeve të sjelljes, trajtimi specifik i fëmijës është i nevojshëm.
Trajtimi i sëmundjes zakonisht është afatgjatë. (1)